Speakers

Professor Jørgen ANDERSEN
Aarhus University, Denmark
Folding of Proteins and RNA Using the Quantum Topology of Moduli Spaces
Abstract
Our attempt at understanding the folding of proteins and RNA using techniques from topology of moduli space will be explained with a quantum twist at the end.

Biography ▼
I. Neural Network Capacity
Professor Pierre BALDI
University of California, USA
Biography
Pierre Baldi earned MS degrees in Mathematics and Psychology from the University of Paris, and a PhD in Mathematics from the California Institute of Technology. He is currently Chancellor's Professor in the Department of Computer Science, Director of the Institute for Genomics and Bioinformatics, and Associate Director of the Center for Machine Learning and Intelligent Systems at the University of California Irvine. The long term focus of his research is on understanding intelligence in brains and machines.
He has made several contributions to the theory of deep learning, and developed and applied deep learning methods for problems in the natural sciences such as the detection of exotic particles in physics, the prediction of reactions in chemistry, and the analysis of circadian rhythms in biology. He has written four books and over 300 peer-reviewed articles.
He is the recipient of the 1993 Lew Allen Award at JPL, the 2010 E. R. Caianiello Prize for research in machine learning, and a 2014 Google Faculty Research Award. He is an Elected Fellow of the AAAS, AAAI, IEEE, ACM, and ISCB.
[ Close ]
I. Neural Network Capacity
II. Systems Biology of Circadian Rhythms
Abstract
First, we will describe a theory of neural network capacity where capacity is defined as the logarithm of the number of functions that can be implemented by a given neural architecture. We will prove theorems showing how capacities can be computed and compared for both shallow and deep architectures. Second, we will provide a comprehensive analysis of multiple high-throughput circadian omic (transcriptomic, proteomic, metabolomic) time series and describe the network-of-coupled-circadian-oscillators theory of circadian rhythms.

Biography ▼
Dr. Brian ATHEY
University of Michigan, USA
Biography
Brian Athey, Ph.D. is the Michael Savageau Collegiate Professor and Founding Chair of the Department of Computational Medicine and Bioinformatics in the U-M Medical School, where he also has appointments as a Professor of Psychiatry and of Internal Medicine. He has previously served as Director of Academic and Clinical Informatics for the School. Brian also serves as Founding Co-director of the U-M wide Michigan Institute for Data Science (MIDAS). Brian is the Principal Investigator of one of the most well established NIH Pre-Doctoral Training Programs in Bioinformatics in the US. He has served as the Principal Investigator on many seminal efforts such as the NIH Visible Human Project, DARPA Virtual Soldier Project, and the NIH National Center for Integrative Biomedical Informatics. He is a founder of the Michigan Institute for Clinical and Health Research (MICHR), where he served for 11 years as Director of the Biomedical Informatics of its Clinical and Translational Science Award (CTSA). He was Co-founder and later CSO of the tranSMART Foundation, a data integration and analytics platform utilized by the pharmaceutical industry globally. Brian’s recent research is the creation and use of bioinformatics pipelines, epigenomics, Electronic Health Records (EHRs), and machine learning methods to radically improve the efficacy of Pharmacogenomics, and is considered a pioneer researcher in the new field of “Pharmacoepigenomics”. From 2010-2016, he served as Chair of Scientific Advisory Board (SAB) of Assurex Health, Inc. (Mason, OH), the leading psychiatric pharmacogenomics company in the world, recently acquired by Myriad Genetics, Inc. (Salt Lake City, UT). He has consulted extensively for the Defense Advanced Research Projects Agency (DARPA), the NIH Office of the Director, the NIH Chief Information Officer. Since 2017, Brian has served as an advisor to the Chinese University Hong Kong, Shenzhen (CUHK-CZ). He is an elected Fellow of the American College of Medical Informatics (FACMI).
[ Close ]
The Emergence of Next Generation Pharmacogenomics Science Enabled by the 4D Nucleome, Patient Data, and Machine Learning Methods
New insights into the architecture and dynamics of the non-coding regulatory genome have transformed our understanding of the cornerstones of classical pharmacogenomics--pharmacokinetics (PK) and pharmacodynamics (PD). The newly emerging concept of the “pharmacoepigenome”, broadly enabled by the 4D Nucleome Concept, contains regulators of gene expression, including enhancers, promoters and RNAs located in the noncoding genome, and is characterized by a hierarchy of stereotypic transcriptional domains in which variation profoundly impacts drug response in humans. Transcriptional control consists of canonical 3D structures that include topologically associated domains (TADs). Different TADs are activated or suppressed in a cell-type specific manner. Drug-disease networks are tightly coupled so that gene variants significantly associated with a disease are identical to, or are found within, the same regulatory networks that determine medication-based therapeutic outcome. Thus, mutations that disrupt the spatial hierarchy of transcription within euchromatin not only convey disease risk but also concomitant variability in drug and pharmacogenomic response. Pathways containing disease risk, drug response and concomitant adverse event variants are better inform therapeutic options for patients based on the emerging pharmacological basis of drug response and adverse drug events. Examples of current and future applications of Machine Learning in pharmacogenomics, include 1) identification of novel regulatory variants located in non-coding domains of the genome and their function as applied to pharmacoepigenomics; 2) patient stratification from medical records; and 3) the mechanistic prediction of drug response, targets and their interactions. In addition, we anticipate that in the future, deep learning will be widely used to predict personalized drug response and optimize medication selection and dosing, using knowledge extracted from large and complex molecular, epidemiological, clinical and demographic datasets.

Biography ▼
Professor Roger BROCKETT
Harvard University, USA
Roger Brockett has contributed widely to control theory and applied mathematics, including early work on linear systems, frequency domain stability theory, differential geometric methods in nonlinear control, the computation of the Volterra series, a geometric approach to the sufficient statistics problem in nonlinear estimation, feedback linearization and feedback stabilization, robot kinematics and dynamics, formal languages for motion control, hybrid systems, computational problems related to tensor ranking and integrable systems, quantum control, and optimal control of Markov processes. His research and teaching has been recognized with awards from the IEEE, ASME, Society for Industrial and Applied Mathematics (SIAM), and AACC and, most recently, International Federation of Automatic Control. He is a Fellow of IEEE, SIAM, and American Mathematical Society and a member of the National Academy of Engineering. As a Harvard faculty member, he served on the faculty council for several years, initiated long-standing introductory-level courses in design and developed major funding for group efforts in robotics and computer vision. He retired from his teaching position in 2012 after nearly 50 years of lecturing while advising more than 60 Ph.D. students at Harvard University, Massachusetts Institute of Technology, and Brandeis University. His 1970 textbook on linear systems has recently been reprinted in the SIAM Classic Series.
[ Close ]
The Role of Heteroclinic Trajectories in Biological Modeling
Abstract
The importance of cyclic, and nearly cyclic processes in mathematical biology is well established and particular examples have been widely studied in a biological context, both as autonomous oscillations and as oscillations under feedback control. On the other hand, there are situations involving growth that seem to be best described as a sequence of episodes which evolve, to some degree, independently of each other. The corresponding mathematical description is less well studied. Using cell division as our main example, we intend to show that differential equations involving multiple heteroclinic equilibria can capture important aspects of episodic growth. Modeling the rise and fall of the concentrations of various members of the Cyclin family of proteins is but one example supporting this claim.

Biography ▼
Professor James Douglas ENGEL
University of Michigan Medical School, USA
Biography
James Douglas Engel’s PhD work in the von Hippel lab was devoted to understanding the thermodynamic behavior of methylated DNA. As a postdoc with Norman Davidson and Tom Maniatis, they generated the first genomic DNA libraries from which we cloned the chicken alpha- and beta-globin loci. They collaboratively were the first to show that DNaseI “hypersensitive sites” in chromatin were coincident with transcriptional regulatory elements. They discovered the first erythroid gene enhancer and subsequently discovered that enhancers function by looping through chromatin to directly interact with promoters. They discovered the GATA transcription factor gene family, and showed that GATA3 loss of function led to embryonic lethality. They later found that conditional GATA3 loss of function led to complete abolition of the T cell lineage. They pioneered the study of mutant transgenic BACs and YACs to study human globin gene regulation and through those studies identified the first gamma-globin repressor. they showed that inactivation of that repressor led to unprescedented induction of fetal globin, a goal for the development of therapeutics to treat sickle cell disease and beta-thallasaemia for several decades. They continue to improve on compounds that exhibit greater gamma-globin induction efficacy.
[ Close ]
GATA3 Regulates Multiple Aspects of Thymocyte Development
Abstract
Thymocytes are unique among hematopoietic cells in that they become developmentally committed only upon maturation in an organ outside the bone marrow. Instructions from different domains of the thymus confer maturational cues that allow thymocytes to first become uniquely committed to the T cell lineage, after which lymphocyte-specific recombination takes place to generate an essentially limitless array of T cell receptors on their surface while simultaneously obeying the rule that each cell must express one and only one antigen receptor. Following several final steps, the selected T cells finally emerge into the bloodstream and become activated after antigen engagement in the periphery. Individual stages of thymocyte maturation can be exquisitely finely monitored by the differential expression of stage-specific antigens during development. One of the key transcription factors that regulates many T cell developmental steps is GATA3. Our studies of the mechanisms by which GATA3 elicits stage-specific developmental responses in thymocytes reveal the complex interplay between multiple aspects of T cell biology: monoallelic gene expression, developmental stage-specific enhancer recruitment and allelic exclusion.

Biography ▼
Professor Leon GLASS
McGill University, Canada
Biography
Leon Glass attended Brooklyn College and did graduate work at the University of Chicago. Before moving to McGill University, where he is currently a Professor of Physiology and the Isadore Rosenfeld Chair in Cardiology, he held postdoctoral positions at the University of Edinburgh, the University of Chicago, and the University of Rochester. He has been a visiting professor at the University of California, San Diego, Harvard Medical School, and Boston University. Dr. Glass is a Fellow of the Royal Society of Canada, the American Physical Society, SIAM, and the American Association for the Advancement of Science. He is the recipient of a Guggenheim Fellowship, the Prix Jacques-Rousseau for interdisciplinary research from ACFAS, and the Arthur T. Winfree Prize from the Society for Mathematical Biology. Dr. Glass' research focuses on the applications of mathematics to study biological function and rhythms in the cardiovascular system, dynamics and control in genetic and neural networks, and visual perception. He is the coauthor (with Michael Mackey) of "From Clocks to Chaos: The Rhythms of Life" (Princeton, 1988), which has been translated into Russian, Chinese and Portuguese, and (with Daniel Kaplan) of "Understanding Nonlinear Dynamics (Springer-Verlag, 1995).
[ Close ]
Geometric Aspects of the Dynamics and Diagnosis of Cardiac Arrhythmias
Abstract
Abnormally rapid heart rhythms, called tachycardias, can be generated by at least two different mechanisms. In one, a localized pacemaker sets a rapid period and entrains some or all of the heart. In the other, the period is set by the time it takes an excitation to travel in some circuitous path. Such rhythms are called reentrant. I will discuss experimental, theoretical and clinical aspects related to predicting patients who are at risk for tachycardia, understanding the mechanisms by which tachycardias are initiated, and treating tachycardias. I will describe various types of reentrant rhythms that are observed in monolayer cardiac cell cultures grown in two geometries that underlie tachycardias: an annulus and an annulus with an isthmus connecting antipodal regions of the annulus. These geometries support reentrant rhythms in which there are one or more circulating waves, as well as rhythms in which there are one or more rotors embedded in the annuli. The experiments and models are relevant to current controversies surrounding mechanisms and treatment of atrial fibrillation - a tachycardia in which the upper chambers of the heart beat irregularly.

Biography ▼
Professor Gemunu GUNARATNE
University of Houston, USA
Biography
Gemunu Gunaratne is a Professor and the Chairman of the Department of Physics at the University of Houston. He received his Ph.D. from Cornell University and conducted post-doctoral research at the University of Chicago before assuming the position at the University of Houston. He proposed the use of periodic orbits and their eigenvalues to characterize strange attractors and showed how dynamical invariants such as fractal dimensions and expansion rates can be derived from them. In spatio-temporal patterns, he invented a class of measures, referred to as the “disorder function,” to quantify the level of pattern irregularity, and used them to describe the dynamics of pattern relaxation. He was a member of the “Chicago” group that first established anomalous scaling properties of hard-turbulence. Gunaratne’s group analyzed “stylized facts” in financial markets, and showed that they follow from a stochastic process with variable diffusion. He has addressed several biologically related problems including establishing linear response as a possible means to establish the strength of osteoporotic bone and developing models to study animal locomotion. More recently, he has focused on model-free control algorithms for coupled networks and turbulent flows.
[ Close ]
Model-Free Network Control
Coupled networks can be used to represent biological processes in cells, interactions within social groups, communications in wireless networks and in many other complex systems. One major goal of network analyses is to design methods to control these systems to pre-specified target states. For example, in cellular processes, we may inquire if damaging consequences of genetic mutations or chromosomal rearrangement can be circumvented through external intervention. The evident approach is to start from a realistic model of the underlying network; unfortunately, this is an extremely difficult task. In intra-cellular processes precise quantitative form of interactions between biomolecules is unlikely to be available. In this talk, I will introduce a model-free approach for control. The “state” of the network is defined using the experimentally accessible gene expression profile. Interestingly, controlling the system to a pre-specified state only requires knowledge of “response surfaces,” which consists of the system responses to a specific set of perturbations. Analyses of model systems and nonlinear electrical circuits show that a target state can be approached by external control of the levels of a few nodes. This approach can prove useful in many contexts including in reprogramming cellular states.

Biography ▼
Professor Alfred HERO
University of Michigan, USA
Biography
Alfred O. Hero III is the John H. Holland Distinguished University Professor of Electrical Engineering and Computer Science and the R. Jamison and Betty Williams Professor of Engineering at the University of Michigan, Ann Arbor. He is also the Co-Director of the University’s Michigan Institute for Data Science (MIDAS). His primary appointment is in the Department of Electrical Engineering and Computer Science and he also has appointments, by courtesy, in the Department of Biomedical Engineering and the Department of Statistics. He received the B.S. (summa cum laude) from Boston University (1980) and the Ph.D from Princeton University (1984), both in Electrical Engineering. He is a Fellow of the Institute of Electrical and Electronics Engineers (IEEE). He has served as President of the IEEE Signal Processing Society and as a member of the IEEE Board of Directors. He has received numerous awards for his scientific research and service to the profession including several best paper awards, the IEEE Signal Processing Society Technical Achievement Award in 2013 and the 2015 Society Award, which is the highest career award bestowed by the IEEE Signal Processing Society. He has received a Rackham Distinguished Faculty Achievement Award in 2011 and the 2017 Stephen S. Attwood Excellence in Engineering Award, from the University of Michigan. Alfred Hero’s recent research interests are in high dimensional spatio-temporal data, multi-modal data integration, statistical signal processing, and machine learning. Of particular interest are applications to social networks, network security and forensics, computer vision, and personalized health.
[ Close ]
Network Inference from Multimodal Genomics
Abstract
We will present a hierarchical generative probabilistic model for inferring the topology of a latent network from multiple modality genomic measurements. This model is applicable to multiple platform assays such as HiC and RNA-seq that are often used to simultaneously probe spatial and functional structure of gene organization. We will review multimodal network analysis methods, develop the proposed hierarchical generative model, describe an optimization approach for inferring the model from data, and illustrate the method by numerical experiments.

Biography ▼
Professor Arthur LANDER
University of California, Irvine, USA
Biography
Dr. Lander enjoys doing “systems” biology, an activity that fuses experimental biology with mathematics, physics, engineering and computer science, in the search for fundamental design principles that explain the complexity of life. He has focused on how cells accurately know their locations in space; how tissues and organs stop growing at precisely-determined sizes; and how selection for control of these processes opens the door to combinatorial fragility, wherein combinations of small changes (e.g. in gene expression) can lead to catastrophic failures (e.g. birth defects).
Dr. Lander received his B.S. degree in Molecular Biophysics and Biochemistry from Yale in 1979, followed by an M.D. degree and a Ph.D. in neuroscience from the University of California, San Francisco in 1985. After postdoctoral research in neurobiology at Columbia University, he joined the faculty of the Massachusetts Institute of Technology in 1987, in the Departments of Brain & Cognitive Sciences and Biology. After receiving tenure, he moved to UC Irvine, where he is currently the Donald Bren Professor of Developmental & Cell Biology, and holds joint appointments in the Departments of Biomedical Engineering and Logic & Philosophy of Science. At UCI, Dr. Lander founded and directs the Center for Complex Biological Systems, designated a National Center for Systems Biology by the NIH, to foster interdisciplinary research, training, and outreach at the interface between biology and the physical, computational, and engineering sciences. He also co-directs UCI’s Cancer Systems Biology Center, and is an Associate Director of the NSF-Simons Center for Multi-scale Cell Fate Research (MathBioSys).
Dr. Lander’s honors include a David and Lucille Packard Fellowship for Science and Engineering; an NIH-Javits Neuroscience Investigator Award; and a RARE Champion of Hope in Science Award, as well as election to membership in the American Society for Clinical Investigation and fellowship in the American Association for the Advance of Science. Dr. Lander has served on the editorial boards of the Journal of Cell Biology, PLoS Biology and BMC Biology, and as a member of multiple NIH grant review panels, including the Modeling and Analysis of Biological Systems Study Section, which he chaired. He is a member of the Clinical Advisory Board and Board of Directors of the Cornelia de Lange Syndrome Foundation, and advocates globally for systems biology through visiting appointments at the University of Tsukuba, in Japan, and National Taiwan University, as well as through teaching in short courses and workshops at various locations in the U.S., Europe and Asia.
[ Close ]
Control of Growth, Form and Scale in Biology
Abstract
Unlike other natural scientists, biologists are obligated to explain not only how phenomena come about (as in “what is the mechanism?”), but also why systems are the way they are (as in “what useful function do they perform?”). Control is broadly defined as those functions that ensure reliable performance in the face of uncertainty. One of the most fascinating arenas in which to study biological control is animal development, because so much happens with high precision, despite massive internal imprecision and unavoidably unpredictable external perturbation. Precision is often achieved in the size of macroscopic structures (tissues and organs), the pattern of cell behavior within those structures, and the relative scale of pattern to size. I will discuss recent data and explicit models of specific feats of invertebrate and vertebrate development to highlight the steep challenges that organisms face in achieving such high control, emphasizing the view that selection for control—even more than selection for performance per se—is likely to have been a major driving force in the evolution of biological complexity.

Dr. Shuai Cheng LI
City University of Hong Kong, Hong Kong
Protein Structures: Space and Dimension
Abstract
Protein Structure area attracted active research. In this talk, we will discuss the modeling of the protein structures. We present a tutorial of protein structures from the aspect of the data sciences and discuss different models of protein structures.

Biography ▼
Dr. Lindsey MUIR
University of Michigan, USA
Biography
In the PhD in molecular and cell biology (University of Washington, Seattle, WA, USA), Lindsey trained in development of gene and cell therapies for skeletal muscle disease, primarily testing the feasibility of using cell reprogramming in autologous therapy that can regenerate skeletal muscle. In the postdoctoral work (University of Michigan, Ann Arbor, MI, USA) Lindsey trained in areas of immunology and metabolism, studying how obesity affects immune cells in mouse and human adipose tissue. In humans, how immune cells such as blood monocytes and tissue macrophages respond in disease settings is yet ill-defined. In postdoctoral and current work at the University of Michigan, Lindsey use high-throughput and genomics technologies with computational tools to quantify and better define these cells and their sub-populations, which may have very different—even opposing—functions. Lindsey also pursue approaches for human immune cell reprogramming that can produce therapeutically beneficial cells in disease settings.
[ Close ]
Genomics of Human Adipose Tissue Macrophages in Obesity
Abstract
Obesity is becoming more prevalent globally, driving a major need to understand the impact of diet and obesity on our health and how it leads to disease. Studies in mice have shown that adipose tissue function is critical to metabolic health. These studies also causally link adipose tissue immune cells, in particular macrophages, to development of insulin resistance in obesity. However, the dynamics and functions of adipose tissue immune cells are not well defined in humans. I will present results on my study of human adipose tissue macrophages from obese patients, their gene expression profiles based on RNA-sequencing data, and comparisons of profiles of three macrophage sub-populations.

Biography ▼
Professor Gilbert OMENN
University of Michigan, USA
Biography
Gil Omenn is the founding director of the Center for Computational Medicine & Bioinformatics and the Harold T. Shapiro Distinguished University Professor of Computational Medicine & Bioinformatics, Internal Medicine, Human Genetics, and Public Health at the University of Michigan, Ann Arbor, MI, USA. His research is focused on cancer proteomics and bioinformatics, especially the role of mRNA and protein splice isoforms, a remarkable development of the evolution of multi-cellular organisms with multi-exon gene structures. He leads the global Human Proteome Project (www.hupo.org). He previously worked on biochemical genetics of the brain, cancer prevention, health promotion and disease prevention for older adults, and science and health policy. He is an author of 592 publications and editor/author of 18 books.
He was a White House Fellow at the U.S. Atomic Energy Commission, Associate Director of the White House Office of Science & Technology Policy and the Office of Management & Budget, and President of the American Association for the Advancement of Science. He is an honorary faculty of Peking Union Medical College and a member of the Peking University/University of Michigan Joint Institute. He advised the Shanghai Jiao Tong University. He participated in the 2016 CUHK IAS Workshop on Mathematics of the Genome. He is an elected member of the National Academy of Medicine, the American Academy of Arts and Sciences, and the American College of Physicians. He received the Walsh McDermott Medal from the National Academy of Sciences and the David Rogers Award from the Association of American Medical Colleges. He holds the BA from Princeton, MD from Harvard, and PhD in Genetics from the University of Washington. He has three children and six grandchildren. He is a musician and tennis player.
Website: www.ccmb.med.umich.edu/omenn
[ Close ]
Computational and Bioinformatic Innovations in Proteogenomics: Predicting Functions of Unannotated Proteins and Facilitating Identification of Missing Proteins
Abstract
Distinguished University Professor of Computational Medicine & Bioinformatics, Internal Medicine, Human Genetics, and Public Health, University of Michigan, Ann Arbor, MI, 48109-2218, USA, and Chair, HUPO Human Proteome Project
Knowledge about proteins is essential for experimental studies and mathematical modeling of the functions of cells and genomes. It is impossible to predict the dynamics and detailed functions of proteins without direct analyses of proteins and their variants, localization, and interactions. For the past 7 years, the Human Proteome Project (HPP) of the Human Proteome Organization (HUPO) has organized global chromosome-centric and biology and disease-driven initiatives to expand and complete the “parts list” of proteins predicted from the human reference genome. Gold standards for progress are the ProteomeXchange of the European Bioinformatics Institute for registering datasets, PeptideAtlas for standardized mass spectrometry analyses, Human Protein Atlas for antibody-based cellular localization of expressed proteins, and neXtProt for curation of protein identifications from all types of biochemical studies. Since 2012, the number of the 20,000 predicted protein-coding human genes for which at least one protein product has been identified using stringent HPP Guidelines has grown from 13,975 to 17,470 (neXtProt PE1 evidence level) proteins, which is 89% of the predicted 19,657. Of these PE1 proteins there are 1260 which still lack specific functional annotations.
Two years ago at the IAS Workshop on Mathematics of the Genome, we discussed detailed findings from our HPP Chromosome 17 team on the effects of alternative splicing of heterogeneous RNAs in dramatically increasing the diversity of proteins (proteoforms), including differential expression of specific splice isoforms in certain cancers. Splicing and the intron/exon structures of genes are dramatic features of the evolution of eukaryotic organisms (above bacteria).
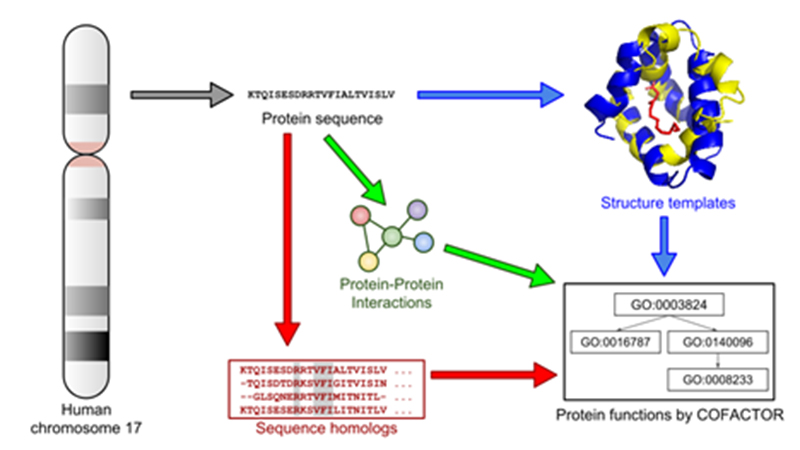
We have just completed a novel analysis of functionally uncharacterized proteins, using the I-TASSER protein structure prediction algorithm followed by the COFACTOR structure-based function annotation algorithm which have been outstanding performers in the worldwide CASP and CAFA challenges. We predict molecular function (MF), biological process (BP), and cellular component (CC) terms of the widely-used Gene Ontology scheme, starting with a benchmarking analysis of 100 randomly-selected well-annotated Chromosome 17 proteins. Then we apply the Cscore thresholds to the corresponding analyses for the 66 uPE1 uncharacterized proteins of Chr 17. We have high-confidence predictions for 13, 33, and 49 proteins by MF, BP, and CC, respectively; using a slightly less stringent set of thresholds we predict specific functions for 30, 39, and 58 of the 66 uPE1 Chr 17 proteins. This is a powerful result from computational analysis, since the challenge to the chromosome-centric HPP teams was to characterize just 2 proteins per chromosome over 2-3 years. This approach can be applied to the entire proteome and can serve as a guide for experimental design and follow-up validation studies.
References: Progress in Identifying and Characterizing the Human Proteome: 2018 (and 2017) Metrics from the Human Proteome Project. Omenn GS, et al, J Proteome Research (submitted) and J Proteome Research 2017, 6:4281-87.
COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 2012; 40: W471-477. Zhang C, Omenn GS, Zhang Y, J Proteome Res (submitted).

Biography ▼
Professor Indika RAJAPAKSE
University of Michigan, USA
Indika Rajapakse is an Associate Professor of Department of Computational Medicine and Bioinformatics, and Department of Mathematics at the University of Michigan, Ann Arbor, MI, USA. Received his B.Sc. degree in electrical engineering and Ph.D. in Applied Mathematics. After postdoctoral work at Fred Hutchinson Cancer Research Center with Mark Groudine joined the faculty at the University of Michigan. His lab is developing genomics and imaging technologies for studying the dynamics of genome organization. A major focus of his lab is to gain a deeper understanding of how the cell cycle guides cell fate determination and also develop strategies for direct reprogramming of normal and abnormal cells. His lab is a part of the Smale Institute. URL: https://rajapakse.lab.medicine.umich.edu/
Algorithm for Cellular Reprogramming
Abstract
I will introduce the concept of data-guided control in building a universal algorithm for directly reprogramming any human cell type into any other type. Our algorithm is based on time series genome transcription and architecture data and known regulatory activities of transcription factors, with natural dimension reduction using genome architectural features (t. Our algorithm predicts known reprogramming factors, top candidates for new settings, and ideal timing for application of transcription factors. This framework can be used to develop strategies for tissue regeneration, cancer cell reprogramming.

Biography ▼
Dr. Thomas RIED
National Cancer Institute, USA
Biography
Dr. Ried received his M.D. degree in 1989 from the Max-Planck-Institute for Medical Research in Heidelberg, Germany, after cloning the b-myosin heavy chain gene and developed the first definitive test for carrier detection of Duchenne muscular dystrophy based on fluorescence in situ hybridization (FISH). Dr. Ried was awarded a three-year postdoctoral fellowship from the German Research Society (Deutsche Forschungsgemeinschaft), which he spent at the Department of Cytochemistry and Cytometry of the University of Leiden, the Netherlands (Dr. van der Ploeg), the Department of Human Genetics, Yale University, New Haven, CT (Dr. David C. Ward), and the Department of Human Genetics of the University of Heidelberg (Dr. Thomas Cremer).
Dr. Ried is one of the pioneers in the development of FISH for the detection of human genetic diseases and cancer, which are exemplified in (i) first in demonstrating that formalin-fixed archived tissue sections can be analyzed using comparative genomic hybridization. He and his colleagues were first in conceptualizing the use of arrayed nucleic acids for comparative genomic hybridization and quantitative DNA and RNA analysis (U.S. Patent # 20030157537). While in Dr. Thomas Cremer’s Laboratory, Dr. Ried was first in demonstrating that formalin-fixed archived tissue sections can be analyzed using comparative genomic hybridization. In fact, this was the first time that retrospective comprehensive screening of tumor genomes became possible, and it precipitated an extensive series of activities in the cancer cytogenetic community in the study of chromosomal aberrations and the correlation of such abnormalities with clinical outcome. Dr. Cremer, Dr. Ried and his colleagues were first in conceptualizing the use of arrayed nucleic acids for comparative genomic hybridization and quantitative DNA and RNA analysis (U.S. Patent # 20030157537). This paved the way for the construction of array-based comparative genomic hybridization and gene expression arrays, tools that are now central to genome research and genetic diagnostics. Dr. Ried’s identification of consistent copy number increases of 3q in cervical cancer was a first. It led to the first patent that was awarded to the National Human Genome Research Institute (then National Center for Human Genome Research, US Patent # 5919624). In 1996, Dr. Ried developed spectral karyotyping, now known more widely by its acronym SKY. He and his colleagues utilized an entirely novel approach for the visualization of FISH experiments. In contrast to conventional epifluorescence filter technology, SKY employs an imaging interferometer linked to a CCD camera to distinguish multiple and overlapping fluorochromes. Today, SKY has been adapted by more than 350 routine cytogenetic diagnostics laboratories all over the world and has become an essential tool for the identification of constitutional and acquired chromosomal aberrations. SKY-based chromosome analysis has been used in close to 1300 publications. Dr. Ried has organized advanced molecular cytogenetic courses at Cold Spring Harbor Laboratories dedicated to educate scientists around the world in this technology.
Dr. Ried’s laboratory now focuses on the role of chromosomal aneuploidy on gene expression in cancer cells, and on how aneuploidy affects nuclear structure. Dr. Ried has published more than 300 peer-reviewed articles, organized two different courses at Cold Spring Harbor Laboratories, serves on numerous editorial boards and as an editor, and as a reviewer for 33 scientific journals and 26 national and international funding agencies. He serves as the Chairman of CancerNet of the German National Genome Research Network, the major funding source for cancer related genome research in Germany. In 2006, Dr. Ried delivered a keynote address at the 20th NIH Intramural Research Festival. Dr. Ried is a member of the steering committee of two Centers of Excellence at the NCI and Associate Professor at Washington University, Washington, D.C., and a Visiting Professor at the University of Göttingen, Germany. In 2000, Dr. Ried received the ASBMB-Amgen Award of the American Society of Biochemistry and Molecular Biology, and was elected an honorary member of the German Society of General and Visceral Surgery in 2009.
[ Close ]
Genome and Transcriptome Dynamics in Cancer Development
Abstract
The development of carcinomas, e.g., cervical or colorectal tumors is defined by the sequential acquisition of chromosomal copy number changes, also referred to as aneuploidy. Importantly, these changes can be invariably characterized as follows: (1) they occur in premalignant lesions before the transition to invasive disease; (2) they are surprisingly tissue specific. In cervical cancers we observe copy number gains of chromosome 3, whereas colon cancers carry extra copies of chromosomes 7, 13 and 20, along with losses of 18; (3) these chromosomal aneuploidies are maintained in metastases and in cell lines, despite ongoing, apparently random, chromosome mis-segregation. Our observations indicate that they are drivers of cancer development and its evolutionary bottleneck. We have conducted extensive analyses to show that genome copy number changes directly affect the expression of genes that reside on the affected chromosomes, both in primary tumors and in experimental models, and therefore result in a complex deregulation of the cancer transcriptome. In addition to chromosomal aberrations, specific genetic signaling pathways are activated by mutations. We are presently analyzing how these cancer specific genetic alterations affect structure/function relationships in the cell nucleus. This is achieved by correlating high-resolution maps of nuclear chromosomal architecture with global gene expression analyses. We realize that such changes cannot be understood in a meaningful way without considering the time aspect in which they occur. We propose applying data guided mathematical methods to understand the dynamical aspect of tumor development, with the ultimate goal of controlling the cancer genome, and reversing cancer cells to the differentiated state they were derived from.
Dr. Hava SIEGELMANN
Defense Advanced Research Projects Agency, USA
Topic to be announced

Biography ▼
Professor Stephen SMALE
City University of Hong Kong, Hong Kong
Biography
Stephen Smale entered the University of Michigan in 1948. Initially, he was a good student, placing into an honors calculus sequence taught by Bob Thrall and earning himself As. However, his sophomore and junior years were marred with mediocre grades, mostly Bs, Cs and even an F in nuclear physics. However, with some luck, Smale was accepted as a graduate student at the University of Michigan's mathematics department. Yet again, Smale performed poorly in his first years, earning a C average as a graduate student. It was only when the department chair, Hildebrandt, threatened to kick Smale out that he began to work hard. Smale finally earned his Ph.D. in 1957, under Raoul Bott.
Smale began his career as an instructor at the college at the University of Chicago. In 1958, he astounded the mathematical world with a proof of a sphere eversion. He then cemented his reputation with a proof of the Poincaré conjecture for all dimensions greater than or equal to 5, published in 1961; in 1962 he generalized the ideas in a 107-page paper that established the h-cobordism theorem.
After having made great strides in topology, he then turned to the study of dynamical systems, where he made significant advances as well. His first contribution is the Smale horseshoe that started significant research in dynamical systems. He also outlined a research program carried out by many others. Smale is also known for injecting Morse theory into mathematical economics, as well as recent explorations of various theories of computation.
[ Close ]
Beating in Unison
Abstract
The myocytes, cells in the heart muscle, organize to coordinate their individual beating into the beating of the heart. The mathematics of this process will be discussed.

Biography ▼
Professor Gary STORMO
Washington University in St. Louis, USA
Biography
Gary Stormo is one of the founders of the field of Computational Biology and has contributed to several areas within that field. He is best known for his work on modeling the specificity of protein-DNA interactions, including the introduction of "weight matrices" to represent short DNA and RNA regulatory motif patterns. His work remains the basis for most motif discovery and analysis programs today. Stormo and his coworkers developed a widely influential formalization of weight matrices in terms of Shannon information theory, providing the mathematical foundation for regulatory motif discovery. He was the first to develop computational methods for discovering motifs in unaligned DNA data, and one of the pioneers of expectation-maximization (EM) algorithms in this field. He did significant subsequent development of this work, improving its statistical footing and increasing its power using comparative genome analysis. His most recent work in this area focuses on obtaining optimal free energy models from high-throughput sequencing data. A related topic was the development of novel methods to predict the specificity of transcription factors based only on their protein sequences.
Stormo was also influential in the fields of RNA structure prediction and protein-coding gene finding. He was the first to take a quantitative approach to RNA structure prediction by comparative sequence analysis based on information theory methods and introduced innovative new methods for RNA folding capable of predicting more complex structures. He also developed methods to predict RNA regulatory motifs that are composed of a combination of sequence and structure constraints. He also introduced methods using dynamic programming techniques to make optimal (highest scoring) predictions of protein coding genes in genome sequences.
In addition to the purely computational work he has invented several new experimental approaches for obtaining quantitative data about protein-DNA interactions, including recent advancements that allow for measurements of the cooperativity between proteins and the effects of epigenetic modifications to the DNA sequences.
Stormo received his BS degree from the California Institute of Technology and his PhD from the University of Colorado. He stayed at the University of Colorado for many years, rising to the rank of Professor, before moving to Washington University School of Medicine in 1999 where he is currently the Joseph Erlanger Professor of Genetics and a member of the Edison Family Center for Genome Sciences and Systems Biology.
[ Close ]
Discovering Multiple Levels of Regulatory Networks
Abstract
This talk will cover our experimental and computational approaches to the study of protein-DNA interactions and specificity and how that influences the regulation of gene expression. Included will be work on cooperativity between transcription factors which plays an important role in vivo in providing specificity. There is also getting to be a greater appreciation for the role of post-transcriptional gene regulation in determining cell state and I will discuss some of our recent work in that area. Besides describing our previous and current work I plan to outline what I think are some of the major open problems related to cellular programming and cell fate decisions.

Biography ▼
Dr. Michael (Xiaohua) XUAN
UniData Technology, China
Biography
Michael (Xiaohua) Xuan is the Founder and Chairman of UniData Technology, a company based in Shanghai, focusing on big data/AI technology and incubation. He also co-founded eBao Technology, an innovative core business system software company for insurance industry. Prior to that, he had worked at Hewlett Packard in USA for eight years in developing large-scale circuit simulation algorithms and commercial software. Michael Xuan holds Ph. D degree in Mathematics from UC Berkeley and MS degree in Computational Mathematics from Zhejiang University in China.
Micahel Xuan is Vice Chairman CSIAM (Chinese Soceity of Inudstrial and Applied Mathematics) and Adjunct Proefessor/member of Academic Committee of Big Data Institute in Fudan University. He is also the executive Director of Smale Institute of Mathematics and Computation.
[ Close ]
Some Applications with Medical Data

Biography ▼
Professor Yuan YAO
The Hong Kong University of Science and Technology, Hong Kong
Yuan Yao received the B.S.E and M.S.E in control engineering both from Harbin Institute of Technology, China, in 1996 and 1998, respectively, M.Phil in mathematics from City University of Hong Kong in 2002, and Ph.D. in mathematics from the University of California, Berkeley, in 2006. Since then he has been with Stanford University and in 2009, he joined as a Fellow of 100-Talent Program the Department of Probability and Statistics in School of Mathematical Sciences, Peking University, Beijing, China. He is currently an Associate Professor of Mathematics, Chemical & Biological Engineering, and by courtesy, Computer Science & Engineering, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China. His current research interests include machine learning and high dimensional data analysis, in particular topological and geometric methods, with applications in computational biology, computer vision, and information retrieval, etc.
[ Close ]
Localization of Heterogeneous Disease Features in Medical Imaging
Identification of disease relevant features has been a crucial challenge in medical image analysis due to the high dimensionality and relative scarcity of patient samples.
Here we introduced some new algorithms developed in our group on disease prediction and important feature discovery for Alzheimer disease and breast cancer detection with imaging data.
The following particular topics will be discussed. A. Equipped with the variable splitting idea and differential inclusion method in optimisation, one can find regularisation paths of parsimonious models that simultaneously exploit both the lesion features and procedure bias to improve the disease prediction; B. To overcome the coherence/colinearity of high dimensional features when imaging resolution increases, an empirical Bayes approach can be exploited with improved accuracy than the multivariate method above for heterogeneous feature discovery; C. Robust statistics can be combined with deep residue learning that help the microcalcification detection in breast cancers.